
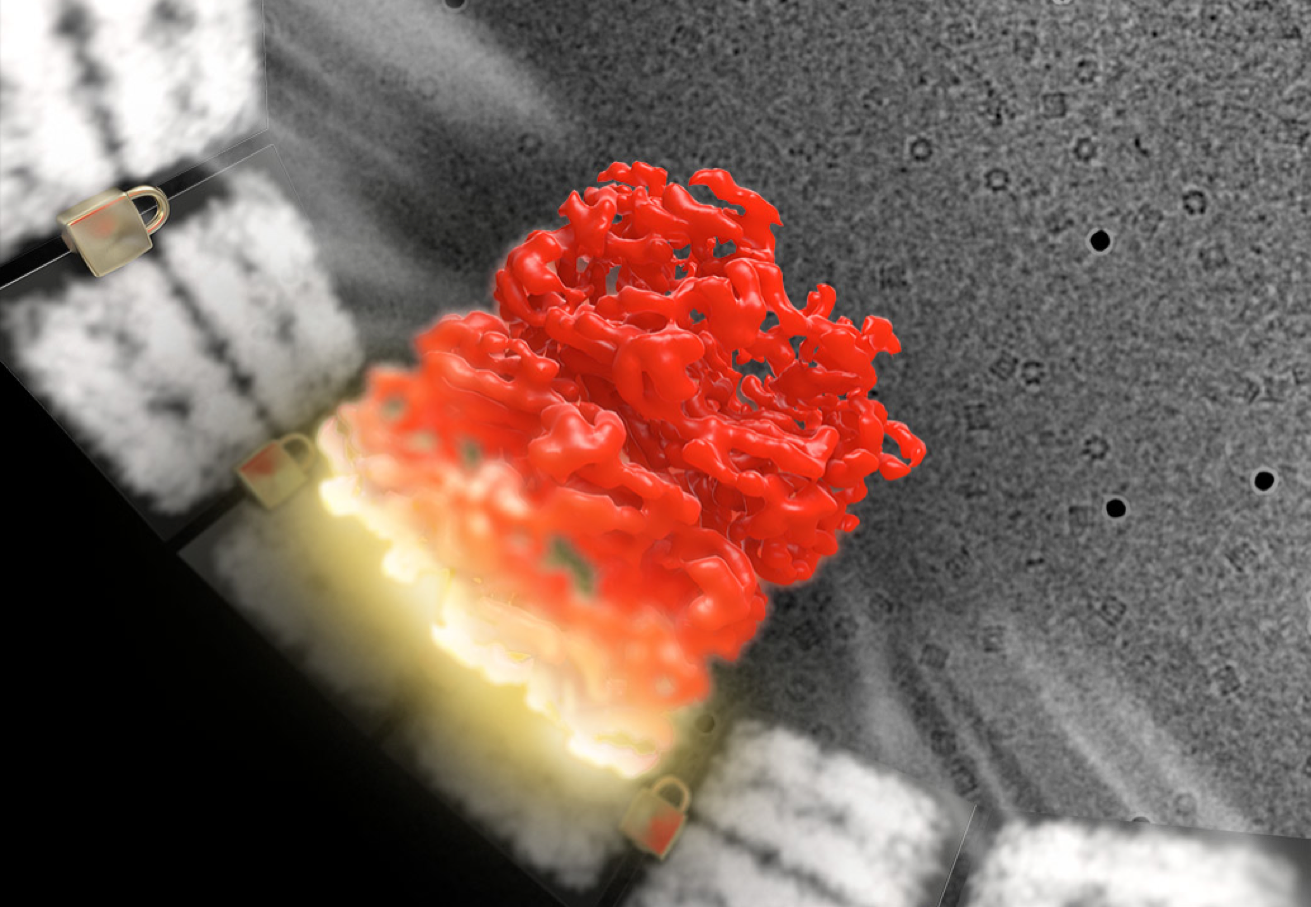


Protein Secondary Structure Determination by Constrained Single-Particle Cryo-Electron Tomography
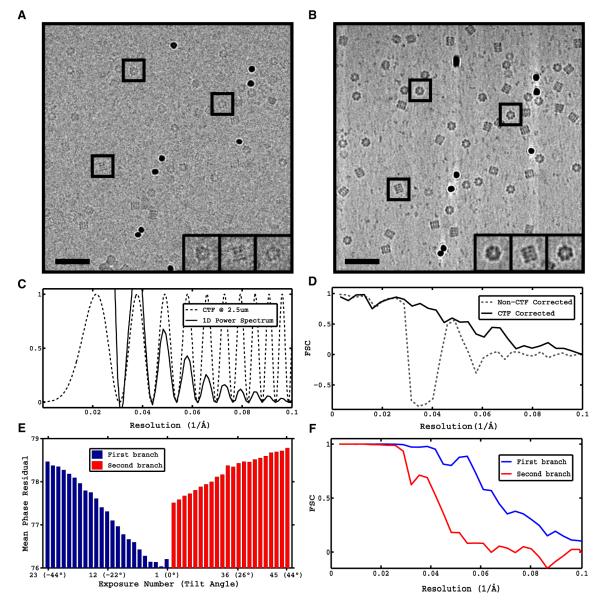
We propose a hybrid strategy, constrained single-particle tomography (CSPT), that combines the advantages of cryo-electron tomography and subvolume averaging with those of traditional single-particle reconstruction and projection matching refinement. Its key components are the effective manipulation of images from tomographic tilt series to successfully extract high-resolution information and correct for the CTF, the incorporation of geometric information provided by tomography into the refinement of image alignments, and improved accuracy of orientation determination by minimizing the effects of model bias and over-refinement associated with working with low-dose tilted projections.
– Efficient extraction of high-resolution information from cryo-EM tilt series.
– Successful CTF correction strategy despite low-contrast and quality of tilted images.
– Use of geometric constraints in refinement improves orientational accuracy of images.
– Enforcement of tomographic constraints reduces model bias and overfitting artifacts.
| Abstract | Cryo-electron microscopy (cryo-EM) is a powerful technique for 3D structure determination of protein complexes by averaging information from individual molecular images. The resolutions that can be achieved with single-particle cryo-EM are frequently limited by inaccuracies in assigning molecular orientations based solely on 2D projection images. Tomographic data collection schemes, however, provide powerful constraints that can be used to more accurately determine molecular orientations necessary for 3D reconstruction. Here, we propose “constrained single-particle tomography” as a general strategy for 3D structure determination in cryo-EM. A key component of our approach is the effective use of images recorded in tilt series to extract high-resolution information and correct for the contrast transfer function. By incorporating geometric constraints into the refinement to improve orientational accuracy of images, we reduce model bias and overrefinement artifacts and demonstrate that protein structures can be determined at resolutions of ∼8 Å starting from low-dose tomographic tilt series. |
|---|