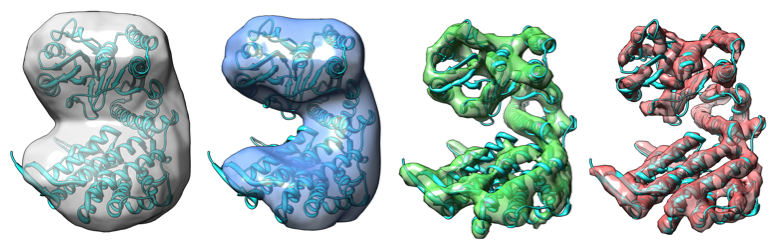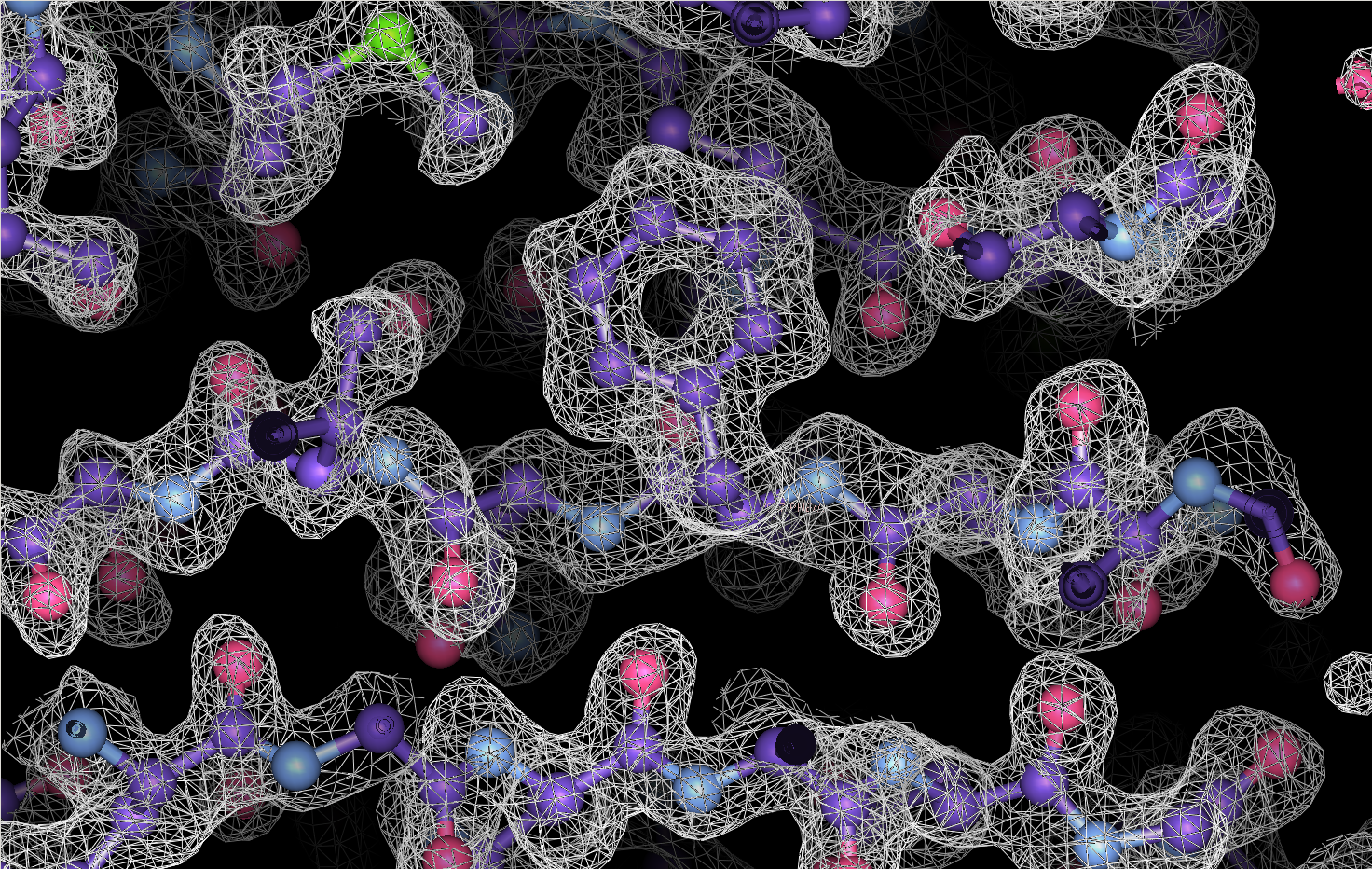
Cryo-EM is a powerful technique for structure determination of protein complexes by averaging information from individual molecular images. Tomographic data collection schemes provide powerful constraints that can be used to more accurately determine molecular orientations necessary for 3D reconstruction. Constrained Single-Particle Tomography is a general strategy for 3D structure determination in cryo-EM that effectively uses images recorded in tilt series to extract high-resolution information and correct for the contrast transfer function of the electron microscope. By incorporating geometric constraints into the refinement of image orientations, we demonstrate that protein structures can be determined at significantly higher resolutions than those obtained using conventional sub-tomogram averaging.